DOCK 5.4 User Manual
Irwin D. Kuntz
Demetri
T. Moustakas
P. Therese Lang
© University of California 2006
Last updated March 2006
General
Overview
Ligand File I/O
Currently, only MOL2 file I/O is supported. Ligands are read in
from a single MOL2 or multi-MOL2 file.
Atom and bond types are assigned using the DOCK 4 atom/bond typing
parameter files (vdw.defn, flex.defn,
flex_table.defn).
More information about all of these files can be found in the
Appendix. There are several ligand output options, which write molecules to files whose
names are formed using the output_file_prefix
parameter:
DOCK
will always write out a scored molecules output file, which contains the best
scoring pose for each molecule in the database.
This will create a file called outputprefix_scored.mol2. Beyond this option, there are several other
levels of sampling output:
1) Users can choose to write out
orientations. This will create a file
called outputprefix_orients.mol2. This
will write out the molecules after they have been rigidly oriented and
optimized. If anchor & grow is being
used, this option will write out only the anchor fragment. All orientations generated will be written out,
so be careful that the output does not get too huge.
2) Users can also write out conformers prior to
final optimization. This will create a
file called outputprefix_confs.mol2.
Again, be aware that the number of molecules in the output file will be
equal to the database size * the # of anchors per molecule * the number of
orientations per anchor * the number of conformers per cycle. This file can grow quite large, so only use
it on single poses or small databases.
3)
Finally, users can write molecules ranked by score. This will create a file called
outputprefix_ranked.mol2, which writes out the top N molecules from the
database. This option disables the
scored molecule output file by default, though users can override this and
write out the best pose for each molecule as well.
Rigid
Orienting
DOCK 5 uses receptor spheres and ligand
heavy atom centers to rigidly orient ligands in the
receptor. The spheres are generated
using the accessory SPHGEN. Cliques of
receptor spheres & ligand centers are identified
using the maximum subgraph clique detection algorithm
from DOCK 4. All cliques that satisfy
the matching parameters are generated in the matching step, and can be sorted
or ordered prior to the loop where the program cycles through the orientations.
Both automated and manual matching are
available in DOCK5. The sphere/center
matches are determined by 2 parameters:
1) The distance tolerance is the tolerance in angstroms
within which a pair of spheres is considered equivalent to a pair of centers
2) The distance minimum is the shortest distance allowed
between 2 spheres (any sphere pair with a shorter distance is disregarded)
Manual
matching will create as many matches as possible given the specified
parameters, and sort the matches according to the RMS error between the spheres
and centers in the match. The matches
are provided as orientations until either the max_orients
# of orientations are reached, or the end of the match list is reached.
Automated matching will start with the default values for
the distance tolerance and distance minimum.
A list of matches will be generated, and if the # of matches is less
than the # max_orientations, then the distance
tolerance is increased and the matching is repeated until there are at least max_orientations in the match list. Then the list is sorted, and orientations are
generated.
Ligand Flexibility
Ligand flexibility in DOCK 5
uses the anchor-and-grow algorithm, which was introduced in DOCK 4. Rotatable bonds
(not contained in rings) are used to partition the molecule into rigid
segments, from which all anchors that meet the criteria are selected beginning
with the largest anchor segment. All
anchor orientations (or the starting orientation only, if no orienting is
selected) are used as starting configurations onto which the first flexible
layer is appended and conformationally expanded. The total population of conformers is then
reduced to the number specified by num_confs_per_cycle,
and the process is repeated until the last layer is reached.
The conformer generator class now integrates score
optimization in the anchor & grow algorithm. The anchors can be rigidly optimized, the
final conformations can be rigidly, torsionally, or
completely optimized, and the partially grown conformers can be completely
optimized. The anchor & grow steps
use whichever scoring function the user selects as the primary scoring
function. The final minimization step
uses the secondary scoring function.
Scoring
Functions
This release of DOCK5 implements a hierarchical scoring
function strategy. A master score class
manages all scoring functions that DOCK uses.
Any of the DOCK scoring functions can be selected as the primary and/or
the secondary scoring function. The
primary scoring function is used during the rigid minimization,
and anchor & grow steps, which typically make many calls to the scoring
function. The secondary scoring function
is used in the final minimization, scoring, and ranking of the molecules. If no secondary scoring function is selected,
the primary scoring function is used as the secondary.
This release contains continuous molecular mechanics
based scoring (vdw + columbic terms only), grid-based
molecular mechanics scoring, contact scoring and bump filtering as implemented
in DOCK 4. Scoring grids are created
using the GRID program. DOCK also
contains GB/SA scoring, as implemented in SDOCK. Scoring grids for the GBSA code are
calculated using the accessories nchemgrid_GB and nchemgrid_SA.
This release also includes an internal energy scoring
function, which is used during the anchor & grow flexible search. This function computes the Lennard-Jones and columbic energy between all ligand atom pairs, excluding all 1-2, 1-3, and 1-4
pairs. This energy is not included in
the final reported score.
Score
Optimization
Score optimization is implemented using a simplex minimizer based on the DOCK 4 minimizer. Users can choose to minimize the rigid
anchors, minimize during flexible growth, and minimize the final
conformation. The anchor minimization is
always done rigidly; also, if no flexible growth is being done, this step will
minimize the entire molecule. The
minimization during the flexible growth is a complete (torsions + rigid)
minimization. The final minimization can
be rigid or torsions only, or complete.
When the simplex “shrinks” enough so that the highest and lowest points
are within the scoring tolerance or if the number of requested minimizer steps is reached, the minimizer
terminates.
Using DOCK
Installing DOCK
1.
Save file for appropriate operating system to hard drive.
2.
Uncompress the archive into a folder called dock5/ in a
directory of your choice.
a.
For windows systems, a Zip file is provided
b.
For *nix systems, a gzipped archive is
provided
3.
All DOCK 5 binaries are installed in dock5/bin/
The dock5 directory contains
the following subdirectories:
bin/
demo/
installation/
parameters/
src/
utilities/
accessories/
grid/
GBSA_Grids/
Compiling DOCK (if required)
DOCK comes with platform
specific compiled binaries. You should
not need to compile the code or accessories unless you have made changes to the
source code, or are planning to run DOCK on a platform for which we do not
distribute binaries.
Building
DOCK: (all platforms)
From the dock5 directory:
cd config/
./configure
gnu
make
DOCK with mpi function is built upon an mpi
library. The MPICH library is provided
freely by Argonne National Labs (http://www-unix.mcs.anl.gov/mpi/mpich/). The MPI library needs to be installed and
running on the system if the MPI features are to be used. Once MPI is installed, you need to define
MPICH_HOME as an environment variable.
Building
MPI-DOCK (all
platforms):
From the dock5 directory:
cd config/
./configure
gnu.parallel
make
NOTE: MPI-DOCK 5.4 has been compiled with
MPICH-1.2.7 on all supported platforms (MPICH-1.2.5 for WinXP).
Running DOCK
For Windows Users:
DOCK and its accessories must be run using a Linux-like environment like Cygwin (http://www.cygwin.com/). When you install your emulator, make sure to also install compilers and unix shells (“Devel” for Cygwin).
DOCK must be run command
line from a standard unix
shell. It reads a parameter file
containing field/value pairs using the following command:
dock5/bin/dock5 -i parameter.in [-v1]
[-v2] [-o outputfile.txt]
If the parameter file does
not exist, DOCK will generate one using your responses to the parameter
questions. If the parameter file exists,
any parameter values found will be read.
DOCK 5 outputs the job
parameters to the screen at the start of the job, and prints summary
information for each molecule processed.
Additional summary information will be included in future releases. The –v1 flag prints a histogram of sphere
matching information. The –v2 flag
prints details about the breakdown of the GB/SA terms.
Running DOCK in Parallel
If you have installed the
MPI library, DOCK can be run in parallel using the following command:
mpirun –np # dock5.mpi -i parameter.in –o outputfile.txt
Note that that parallezation is set up to have a single Master node with
the remaining nodes act as slaves. The
Master node performs file processing and input/output, whereas the slaves
perform the actual calculations. If –np = 1, the code defaults to non-MPI behavior. As a result, there will be minimal difference
in performance between 1 and 2 processors.
Improved performance will only become evident with more than 2 nodes.
Running the Demo
DOCK 5.4 includes two demonstration
files that are designed to test your installation. These demos must also be run command-line.
For
DOCK: (all platforms)
From the dock5 directory:
cd demo
./script_clean
./script_demo
For MPI-DOCK: (all platforms)
From the dock5 directory:
cd demo
./script_clean
./script_mpi_demo
NOTE: MPI-DOCK
will be run on 4 processors for the demo
DOCK 5 Parameters
The parameters for several
common calculations have been optimized using test sets. General recommendations for these parameters
can be found in dock5/recommended_input.
Below, all available options for DOCK are described in detail.
The DOCK 5 parameter parser
requires that the values entered for a parameter exactly match one of the legal
values if any legal values are specified.
For example:
param_a [5] ():
param_b [5] (0 5 10):
param_a can be
assigned any value, however param_b can only be
assigned 0, 5, or 10. If no value is
entered, both will default to a value of 5.
Below are listed all DOCK 5 parameters, their default values, legal
values, and a brief description of each.
The parameters are listed in order of function. Also, for questions requiring a yes/no
answer, please use the full word (yes or no) as opposed to y or n.
Ligand I/O Parameters
|
Parameter Name |
Default |
Values |
Description |
|
ligand_atom_file |
database.mol2 |
string |
The ligand input filename |
|
ligand_outfile_prefix |
output |
string |
The
prefix that all output files will use |
|
limit_max_ligands |
no |
bool (yes, no) |
The
maximum # of ligands that will be read in from a
library |
|
write_orientations |
no |
bool (yes, no) |
Flag to
write orientations |
|
write_conformations |
no |
yes, no |
Flag to
write conformations |
|
initial_skip |
0 |
int |
The # of
molecules to skip over at the beginning of a library |
|
calculate_rmsd |
no |
yes, no |
Flag to
perform an RMSD calculation between the final molecule pose and its initial
structure. |
|
use_rmsd_reference_mol |
no |
yes, no |
Specify reference structure for RMSD calculation (default is starting structure) |
|
rmsd_reference_filename |
ligand_rmsd.mol2 |
string |
File containing RMSD reference structure |
|
rank_ligands |
no |
yes, no |
Flag to
enable a ligand top-score list. These ligands
will be written to outfile_ranked.mol2, and outfile_scored.mol2 will be empty
by default |
|
max_ranked_ligands |
500 |
int |
The # of ligands to be stored in the top score list |
|
scored_conformer_output_override |
no |
yes, no |
This flag
causes all ligands to be written to
outfile_scored.mol2, even when rank_ligands is true |
|
num_scored_conformers_written |
1 |
int |
The # of
scored poses for each ligand printed to
output_scored.mol2 |
|
cluster_conformations |
yes |
yes, no |
Flag to
enable clustering of fully minimized conformations |
|
cluster_rmsd_threshold |
2.0 |
float |
The
cutoff to determine whether conformations should be clustered |
Orient Ligand Parameters
|
Parameter Name |
Default |
Values |
Description |
|
orient_ligand |
yes |
bool (yes, no) |
Flag to
orient ligand to spheres |
|
automated_matching |
yes |
bool (yes, no) |
Flag to
perform automated matching instead of manual matching |
|
distance_tolerence |
0.25 |
float |
The
distance tolerance applied to each edge in a clique |
|
distance_minimum |
2.0 |
float |
The
minimum size for an edge in a clique |
|
nodes_minimum |
3 |
int |
The
minimum # of nodes in a clique |
|
nodes_maximum |
10 |
int |
The
maximum # of nodes in a clique |
|
receptor_site_file |
receptor.sph |
string |
The file
containing the receptor spheres |
|
max_orientations |
500 |
int |
The
maximum # of orientations that will be cycled through |
|
critical_points |
no |
bool (yes, no) |
Flag to use
critical point sphere labeling to target orientations to particular spheres |
|
chemical_matching |
no |
bool (yes, no) |
Flag to
use chemical “coloring” of spheres to match chemical labels on ligand atoms |
|
chem_match_tbl |
chem_match.tbl |
string |
File
defining the legal chemical type matches/pairings |
|
use_ligand_spheres |
no |
bool (yes, no) |
Flag to
enable a sphere file representing ligand heavy
atoms to be used to orient the ligand. Typically used for macromolecular docking |
|
ligand_sphere_file |
ligand.sph |
string |
Ligand spheres |
Flexible Ligand Parameters
|
Parameter Name |
Default |
Values |
Description |
|
flexible_ligand |
yes |
bool (yes, no) |
Flag to
perform ligand conformational searching |
|
ag_conf_search |
yes |
bool (yes, no) |
Flag to
use the anchor & grow algorithm to search ligand
conformations |
|
min_anchor_size |
40 |
int |
The
minimum # of heavy atoms for an anchor segment |
|
num_anchor_orients_for_growth |
100 |
int |
The
maximum number of anchor orientations promoted to the conformational search |
|
number_confs_for_next_growth |
100 |
int |
The
maximum number of conformations carried forward in the anchor & grow
search |
|
use_internal_energy |
yes |
bool (yes, no) |
Flag to add
an internal energy term to the score during the conformational search |
|
internal_energy_att_exp |
6 |
int |
VDW
attractive exponent |
|
internal_energy_rep_exp |
12 |
int |
VDW
repulsive exponent |
|
internal_energy_dielectric |
4.0 |
float |
Dielectric
used for electrostatic calculation |
|
use_clash_overlap |
no |
bool (yes, no) |
Flag to
check for overlapping atom volumes during anchor and grow |
|
clash_overlap |
0.5 |
float |
Percent
of overlap allowed before a clash is declared |
Ligand Scoring Parameters
|
Parameter Name |
Default |
Values |
Description |
|
bump_filter |
yes |
bool (yes, no) |
Flag to
perform bump filtering |
|
bump_grid_prefix |
grid |
string |
The
prefix to the grid file(s) containing the desired bump grid |
|
max_bumps |
2 |
int |
The
maximum allowed # of bumps for a molecule to pass the filter |
|
score_molecules |
yes |
bool (yes, no) |
Enables
scoring of molecules |
|
contact_score_primary |
no |
bool (yes, no) |
Flag to
perform contact scoring as the primary scoring function |
|
contact_score_secondary |
no |
bool (yes, no) |
Flag to
perform contact scoring as the secondary scoring function |
|
contact_score_cutoff_distance |
4.5 |
float |
The
distance threshold defining a contact |
|
contact_score_clash_overlap |
0.75 |
float |
Contact
definition for use with intramolecular scoring |
|
contact_score_clash_penalty |
50 |
int |
The
penalty for each contact overlap made |
|
contact_score_grid_prefix |
grid |
string |
The
prefix to the grid files containing the desired cnt
grid |
|
grid_score_primary |
yes |
bool (yes, no) |
Flag to perform grid-based energy scoring as the primary scoring function |
|
grid_score_secondary |
yes |
bool (yes, no) |
Flag to perform grid-based energy scoring as the secondary scoring function |
|
grid_score_vdw_scale |
1 |
float |
Scalar multiplier of the vdw energy component |
|
grid_score_es_scale |
1 |
float |
Scalar multiplier of the electrostatic energy component |
|
grid_score_grid_prefix |
grid |
string |
The prefix to the grid files containing the desired nrg grid |
|
continuous_score_primary |
no |
bool (yes, no) |
Flag to perform continuous non-grid based scoring |
|
continuous_score_secondary |
no |
bool (yes, no) |
Flag to perform continuous non-grid based scoring |
|
cont_score_rec_filename |
receptor.mol2 |
string |
File that contains receptor coordinates |
|
cont_score_att_exp |
6 |
int |
VDW L-J potential attractive exponent |
|
cont_score_rep_exp |
12 |
int |
VDW L-J potential repulsive exponent |
|
cont_score_dielectric |
4.0 |
float |
Dielectric constant for electrostatic term |
|
cont_score_vdw_scale |
1 |
float |
Scalar multiplier of vdw energy component |
|
cont_score_es_scale |
1 |
float |
Scalar multiplier of electrostatic energy component |
|
gbsa_pairwise_score_primary |
no |
bool (yes, no) |
Toggles
whether or not to use GB/SA scoring as the primary scoring function |
|
gbsa_pairwise_score_secondary |
no |
bool (yes, no) |
Toggles
whether or not to use GB/SA scoring as the secondary scoring function |
|
gbsa_pairwise_gb_grid_prefix |
gb_grid |
string |
The path
to the pairwise GB grids |
|
gbsa_pairwise_sa_grid_prefix |
sa_grid |
string |
The path
to the SA grids |
|
gbsa_pairwise_vdw_grid_prefix |
grid |
string |
The path
to the dock4 nrg grids, used for the vdw portion of the GB/SA calculation |
|
gbsa_pairwise_screen_file |
screen.in |
string |
GB parameter file for electrostatic screening. Its located in the parameters dir by
default |
|
gbsa_pairwise_solvent_dielectric |
78.300003 |
float |
The value
for the solvent dielectric |
Score Optimization Parameters
|
Parameter Name |
Default |
Values |
Description |
|
minimize_ligand |
yes |
bool (yes, no) |
Flag to
perform score optimization |
|
minimize_anchor |
yes |
bool (yes, no) |
Flag to
perform rigid optimization of the anchor |
|
minimize_flexible_growth |
yes |
bool (yes, no) |
Flag to perform
complete optimization during conformational search |
|
minimize_final_pose |
yes |
bool (yes, no) |
Flag to
perform minimization of the final ligand pose |
|
use_advanced_simplex_parameters |
no |
bool (yes, no) |
Flag to
use a simplified set of common minimization parameters for each of the
minimization steps listed above |
Basic Simplex Minimizer Parameters
|
Parameter Name |
Default |
Values |
Description |
|
simplex_max_cycles |
1 |
int |
Maximum #
of minimization cycles |
|
simplex_score_converge |
0.1 |
float |
Exit
cycle at when energy converges at cutoff |
|
simplex_cycle_converge |
1.0 |
float |
Exit
minimization when cycles converge at cutoff |
|
simplex_trans_step |
1.0 |
float |
Initial
translation step size |
|
simplex_rot_step |
0.1 |
float |
Initial
rotation step size |
|
simplex_tors_step |
10.0 |
float |
Initial
torsion angle step size |
|
simplex_anchor_max_iterations |
50 |
int |
Maximum #
of iterations per cycle per anchor |
|
simplex_grow_max_iterations |
500 |
int |
Maximum #
of iterations per cycle per growth step |
|
simplex_final_max_iterations |
0 |
int |
Maximum #
of iterations per cycle for entire molecule |
|
simplex_random_seed |
0 |
int |
Seed for
random number generator |
Advanced Simplex Minimizer Parameters
|
Parameter Name |
Default |
Values |
Description |
|
simplex_anchor_max_iterations |
50 |
int |
Maximum #
of minimization cycles |
|
simplex_anchor_max_cycles |
1 |
int |
Maximum #
of minimization cycles |
|
simplex_anchor_score_converge |
0.1 |
float |
Exit
cycle at when energy converges at cutoff |
|
simplex_anchor_cycle_converge |
1.0 |
float |
Exit
minimization when cycles converge at cutoff |
|
simplex_anchor_trans_step |
1.0 |
float |
Initial
translation step size |
|
simplex_anchor_rot_step |
0.1 |
float |
Initial
rotation step size |
|
simplex_anchor_tors_step |
10.0 |
float |
Initial
torsion angle step size |
|
NOTE: Repeat
for minimization of each layer (simplex_grow_xxx)
and final minimization (simplex_final_xxx)
|
|||
Atom & Bond Typing Parameters
|
Parameter Name |
Default |
Legal |
Description |
|
atom_model |
all |
string
(all, united) |
Choice of
all atom or united atom models |
|
vdw_defn_file |
vdw.defn |
string |
File
containing vdw parameters for atom types |
|
flex_defn_file |
flex.defn |
string |
File
containing bond definition parameters |
|
flex_drive_file |
flex_drive.tbl |
string |
File
containing conformational search parameters |
|
chem._defn_file |
chem.defn |
string |
File
containing chemical label (pharmacophore)
definitions |
ACCESSORIES
GRID
Author:
Todd Ewing
Based on
work by Elaine Meng and Brian Shoichet
Overview
GRID creates
the grid files necessary for rapid score evaluation in DOCK. Two types of
scoring are available: contact and energy scoring. The scoring grids are stored
in files ending in *.cnt and *.nrg
respectively. When docking, each scoring function is applied independent of the
others and the results are written to separate output files. GRID also computes a bump grid which
identifies whether a ligand atom is in severe steric overlap with a receptor atom. The bump grid is
identified with a *.bmp file extension. The file containing the bump grid also
stores the size, position and grid spacing of all the grids.
The grid
calculation must be performed prior to docking. The calculation can take up to
45 minutes, but needs to be done only once for each receptor site. Since DOCK
can perform continuum scoring without a grid, the grid calculation is not
always required. However, for most docking tasks, such as when multiple binding
modes for a molecule or multiple molecules are considered, it will become more
time efficient to precompute the scoring grids.
Bump Checking
Prior to
scoring, each orientation can be processed with the bump filter to reject ones
that penetrate deep into the receptor. Orientations that pass the bump filter
are then scored and/or minimized with any of the available scoring
functions. A bump is based on the sum of
the van der Waals radii of
the two interacting atoms. The user specifies what fraction of the sum is
considered a bump. For example, the default definition of a bump is if any two
atoms approach closer than 0.75 of the sum of their radii. GRID stores an atomic radius which
corresponds to smallest radius of ligand atom at the
grid position which would still trigger a bump. During docking, for a given
orientation, the position of each atom is checked with the bump grid. If the
radius of the atom is greater than or equal to the radius stored in the bump
grid, then the atom triggers a bump. To conserve disk space, the atom radius is
multiplied by 10 and converted to a short unsigned integer.
Energy scoring
The energy
scoring component of DOCK is based on the implementation of force field
scoring. Force field scores are approximate molecular mechanics interaction
energies, consisting of van der Waals
and electrostatic components:
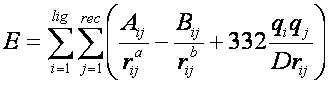
75
where each term is a double sum over ligand atoms i and receptor atoms
j, which include the quantities listed below.
Generalization
of the VDW component
The van der Waals component of the
scoring function has been generalized to handle any combination of repulsive
and attractive exponents (providing that a> b). The user may choose to
"soften" the potential by using a 6-9 Lennard-Jones
function. The general form of the van der Waals interaction between two identical atoms is presented:
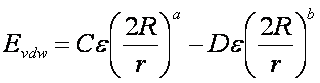
where e is the well depth of the interaction
energy, R is the van der Waals
radius of the atoms, and coefficients C and D can be determined given the two
following boundary conditions:
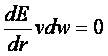 at
at ![]()
![]() at
at![]()
Application of these boundary conditions
to the above equation yields an expression of
the van der Waals
interaction with a generalized Lennard-Jones
potential.
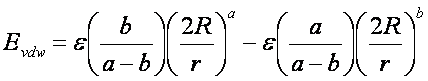
The consequence of using a different
exponent for the repulsive term is illustrated in Figure 1. Notice that the
well position and depth are unchanged, but that the repulsive barrier has
shrunk by about a 0.25 Angstrom.
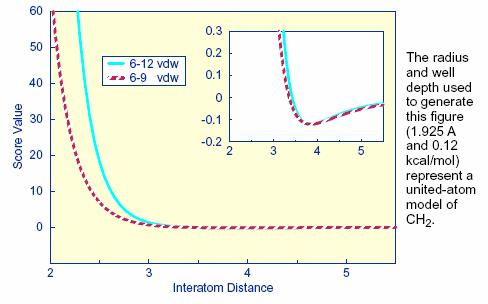
Figure
1: Distance dependence of the Lennard-Jones Function
Precomputing potentials on a grid
By inspection
of the above equations, the repulsion and
attraction parameters (Aij and Bij)
for the interactions of identical atoms can be derived from the van der Waals radius, R, and the well
depth, e.
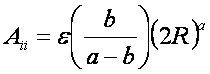
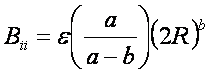
In order to evaluate the interaction
energy quickly, the van der Waals
and electrostatic potentials are precomputed for the
receptor and stored on a grid of points containing the docking site. Precomputing the van der Waals potential requires the use of a geometric mean
approximation for the A and B terms, as shown:
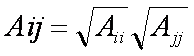
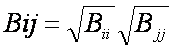
Using this approximation, the first equation can be rewritten:
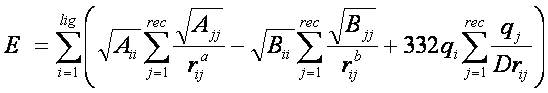
Three values are stored for every grid
point k,each a sum over
receptor atoms that are within a user defined cutoff distance of the point:


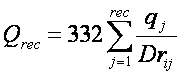
These values, with trilinear
interpolation, are multiplied by the appropriate ligand
values to give the interaction energy. GRID calculates the grid values and
stores them in files. The values are read in during a DOCK run and used for
force field scoring.
The user
determines the location and dimensions of the grid box using the program SHOWBOX. It is not necessary for the whole receptor to be enclosed;
only the regions where ligand atoms may be placed
need to be included. The box merely delimits the space where grid points are
located, and does not cause receptor atoms to be excluded from the calculation.
Besides a direct specification of coordinates, there is an option to center the
grid at a sphere cluster center of mass. Any combination of spacing and x, y,
and z extents may be used.
Contact Scoring
Contact scoring in GRID incorporates the
scoring performed with the DISTMAP program developed by Shoichet
and Bodian. The score is a summation of the heavy
atom contacts (every atom except hydrogen) between the ligand
and receptor. A contact is defined as an approach of two atoms within some
cutoff distance (usually 4.5 Angstroms). If the two atoms approach
close enough to bump (as identified with the bump grid) then the
interaction can be penalized by an amount specified by the user. The distance
dependence of the contact score is represented in Figure 2.
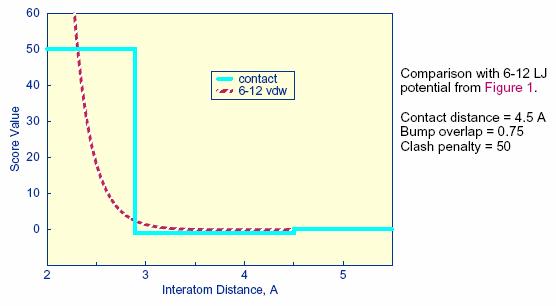
Figure 2. Distance
dependence of contact score function
The attractive score in GRID is negative
and a repulsive score is positive. This
switch of sign is necessary to allow the same minimization protocol to be used
for contact scoring as implemented for energy scoring.
Running GRID
GRID
must be run command line from a standard unix
shell. It reads a parameter file
containing field/value pairs using the following command:
grid -i
grid.in [-stv] [-o grid.out]
-i input_file (Input parameters
extracted from input_file, or grid.in
if not specified)
-o output_file (Output written to output_file,
or grid.out if not specified)
-s Input parameters
entered interactively
-t Reduced
output level
-v Increased
output level
General Parameters
|
Parameter Name |
Default |
Value |
Description |
|
compute_grids |
no |
bool (yes, no) |
Flag to compute
scoring grids |
|
grid_spacing |
0.3 |
float |
The distance between
grid points along each axis. |
|
output_molecule |
no |
bool (yes, no) |
Flag to write out the
coordinates of the receptor into a new, cleaned-up file. Atoms are resorted
to put all residue atoms together. Terminal SYBYL capping groups are merged
with the terminal residues. |
Scoring Parameters
|
Parameter Name |
Default |
Value |
Description |
|
contact_score |
no |
bool (yes, no) |
Flag to construct
contact grid |
|
contact_cutoff_distance |
4.5 |
float |
Maximum distance between
heavy atoms for the interaction to be counted as a contact |
|
chemical_score |
no |
bool (yes, no) |
Flag to construct
chemical grid |
|
energy_score |
no |
bool (yes, no) |
Flag to perform energy
scoring |
|
energy_cutoff_distance |
10 |
float |
Maximum distance
between two atoms for their contribution to the energy score to be computed |
|
atom_model |
u |
string (u, a) |
Flag for how to model
of nonpolar hydrogens u = United atom model. Hydrogens attached to carbons are assigned a zero VDW well-depth and the partial charge is transferred to the carbon. a = All atom model. Hydrogens attached to carbons have regular VDW well-depth and
partial charge is not modified. |
|
attractive_exponent |
6 |
int |
Exponent of attractive
Lennard- Jones term for VDW potential |
|
repulsive_exponent |
12 |
int |
Exponent of repulsive Lennard-Jones term for VDW potential |
|
distance_dielectric |
yes |
bool (yes, no) |
Flag to make the
dielectric depend linearly on the distance |
|
dielectric_factor |
4.0 |
float |
Coefficient of the
dielectric |
|
bump_filter |
no |
bool (yes, no) |
Flag to screen each
orientation for clashes with receptor prior to scoring and minimizing |
|
bump_overlap |
0.75 |
float |
Amount of VDW overlap allowed.
If the probe atom and the receptor heavy atom approach closer than this
fraction of the sum of their VDW radii, then the position is flagged as a
bump. 0 = Complete overlap
allowed. 1 = No overlap allowed |
File Input
|
Parameter Name |
Default |
Value |
Description |
|
receptor_file |
receptor.mol2 |
bool (yes, no) |
Receptor coordinate
file. Partial charges and atom types need to be present. |
|
box_file |
site_box.pdb |
float |
File containing SHOWBOX output
file which specifies boundaries of grid |
|
vdw_definition_file |
vdw.defn |
string |
VDW parameter file. |
File Output
|
Parameter Name |
Default |
Value |
Description |
|
score_grid_prefix |
grid |
string |
Core file name of grids
(file extension will be appended automatically) |
|
receptor_out_file |
receptor_out.mol2 |
string |
File for cleaned-up
receptor when output_molecule set |
NCHEMGRID_GB and NCHEMGRID_SA
Author: Xiaoqin Zou
Overview
The NCHEMGRID_GB and NCHEMGRID_SA programs create the
GB and SA receptor grids for use with DOCK 5's GB/SA scoring function.
Input
Both programs require that
an INCHEM file be created in the working directory, which contains the
parameters to control the program. The
INCHEM parameters for both the NCHEMGRID_GB and NCHEMGRID_SA programs are
detailed below:
For NCHEMGRID_GB:
receptor.pdb ; receptor pdb file
cavity.pdb ; cavity pdb file
parameters/prot.table.ambcrg.ambH ; charge parameter file
parameters/vdw.parms.amb ; VDW parameter file
box.pdb ; box pdb file
0.4 ; grid
spacing in angstroms
2
; es type: GB
1
; es scale for ff
scoring
8.0 8.0 ; cutoff for es and
outer box
78.3 78.3 ; dielectric of solvent,cavity
2.3 2.8 ; bumping distances
output_prefix ; output grid prefix name
1 ; pairwise
calculation
NOTE: The cavity.pdb file should be an empty file. This feature is not frequently used. However, the parameter must still be
passed. The pairwise
calculation value must also always be 1.
For NCHEMGRID_SA:
receptor.pdb ; receptor pdb file
pararameters/prot.table.ambcrg.ambH ;
charge parameter file
parameters/vdw.parms.amb ; VDW parameter file
box.pdb
;
box pdb file
0.4 ; grid spacing in angstroms
1.4 ; probe radius for SA
2 ; scoring type: SA
8.0 ; cutoff for SA calculations
output_prefix ; output grid prefix name
SPHGEN
Author:
Irwin D. Kuntz
Modified by:
Renee DesJarlais, Brian Shoichet
Overview
SPHGEN
generates sets of overlapping spheres to describe the shape of a molecule or
molecular surface. For receptors, a negative image of the surface invaginations
is created; for a ligand, the program creates a
positive image of the entire molecule. Spheres are constructed using the
molecular surface described by Richards (1977) calculated with the program dms (www.cgl.ucsf.edu). Each sphere touches the molecular surface at two points
and has its radius along the surface normal of one of the points. For the
receptor, each sphere center is “outside” the surface, and lies in the
direction of a surface normal vector. For a ligand, each
sphere center is “inside” the surface, and lies in the direction of a reversed
surface normal vector. Spheres are calculated over the entire surface,
producing approximately one sphere per surface point. This very dense
representation is then filtered to keep only the largest sphere associated with
each receptor surface atom. The filtered set is then clustered on the basis of
radial overlap between the spheres using a single linkage algorithm. This
creates a negative image of the receptor surface, where each invagination is characterized by a set of overlapping
spheres. These sets, or “clusters,” are sorted according to numbers of
constituent spheres, and written out in order of descending size. The largest
cluster is typically the ligand binding site of the
receptor molecule. The program showsphere writes out sphere center coordinates in PDB format and may
be helpful for visualizing the clusters.
Input
rec.ms #molecular surface
file
R #sphere outside of
surface (R) or inside surface (L)
X #specifies
subset of surface points to be used (X=all points)
0.0 #prevents
generation of large spheres with close surface contacts (default=0.0)
4.0 #maximum
sphere radius in Angstroms (default=4.0)
1.4 #minimum
sphere radius in Angstroms (default=radius of probe)
rec.sph #clustered spheres file
NOTES:
1) The input file names and parameters are read
from a file called INSPH, which should not contain any blank lines or the
comments (denoted by #) from above.
2) The molecular surface file must include surface
normals. SPHGEN expects the Fortran
format
(A3, I5, X, A4, X, 2F8.3, F9.3, X, A3, 7X, 3F7.3).
Output
Some
informative messages are written to a file called OUTSPH. This includes the
parameters and files used in the calculation. The spheres themselves are
written to the clustered spheres file. They are arranged in clusters with the
cluster having the largest number of spheres appearing first. The sphere
cluster file consists of a header followed by a series of sphere clusters. The
header is the line “DOCK 3.5 receptor_spheres”
followed by a color table. The color table contains color names (format A30)
each on a separate line. As SPHGEN
produces no colors, the color table is simply absent.
The sphere
clusters themselves follow, each of which starts with the line
cluster
n number of spheres in cluster i
where n is the cluster number for that sphere
cluster, and i is the number of spheres in that
cluster. Next, all spheres in that
cluster are listed in the format (I5, 3F10.5, F8.3, I5, I2, I3) where the values correspond to, respectively,
- The number of the atom with which surface point i
(used to generate the sphere) is associated.
- The x, y,and z
coordinates of the sphere center.
- The sphere radius.
- The number of the atom with which surface point j (second point
used to generate the sphere) is associated.
- The critical cluster to which this sphere belongs.
- The sphere color. The color is simply an index into the color table
that was specified in the header. Therefore, 1 corresponds to the first
color in the header, 2 for the second, etc. 0 corresponds to
unlabeled.
The clusters
are listed in numerical order from largest cluster found to the smallest. At
the end of the clusters is cluster number 0. This is not an actual sphere
cluster, but a list of allof the spheres generated
whose radii were larger than the minimum radius, before the filtering
heuristics ( i.e. allowing only one sphere per atom
and using a maximum radius cutoff) and clustering were performed. Cluster 0 may
be useful as a starting point for users who want to explore a wider range of
possible clusters than is provided by the standard SPHGEN clustering
routine. The program creates three
temporary files: temp1.ms, temp2.sph, and temp3.atc. These are used internally
by SPHGEN, and are deleted upon completion of the computation. For more information on sphere generation and
selection, go to the demo.
SHOWBOX
Author:
Elaine Meng
SHOWBOX is an interactive program that
allows visualization of the location and size of the grids that will be
calculated by the program grid, using any graphics program that can
display PDB format. The user is asked whether the box should be automatically
constructed to enclose all of the spheres in a cluster. If so, the user must
also enter a value for how closely the box faces may approach a sphere center
(how large a “cushion” of space is desired) and the sphere cluster filename and
number. If not, the user is asked whether the box will be centered on manually
entered coordinates or a sphere cluster center of mass. Depending on the
response, the coordinates of the center or the sphere cluster filename and
number are requested. Finally, the user must enter the desired box dimensions
(if not automatic) and a name for the output PDB-format box file.
SHOWSPHERE
Authors:
Stuart Oatley, Elaine Meng,
Daniel Gschwend
SHOWSPHERE is an interactive program; it
produces a PDB-format file of sphere centers and an MS-like file of sphere
surfaces, given the sphere cluster file and cluster number. The surface file
generation is optional. The user may
specify one cluster or “all,” and multiple output files will be generated, with
the cluster number appended to the end of the name of each file. The input
cluster file is created using SPHGEN. SHOWSPHERE requests the name of the sphere
cluster file, the number of the cluster of interest, and names for the output
files. Information is sent to the screen while the spheres are being read in,
and while the surface points are being calculated.
SPHERE_SELECTOR
Author: P.
Therese Lang
SPHERE_SELECTOR will take the ouput from SPHGEN and select all spheres with a
user-defined radius of a target molecule.
The target molecule can be anything (ie known ligand, receptor residue, ect) as long as it is in proper MOL2 format. The required input for sphere_selector
is:
USAGE: sphere_selector <sphere_cluster_file.sph>
<set_of_atoms.mol2> <radius>
Please note that above order of
input files must be maintained for the program to work.
APPENDIX 1: Parameter File Formats
Overview
The parameter
files contain atom and bond data needed during DOCK calculations. The
definition (*.defn) files contain atom and bond
labeling data. The table (*.tbl) files contain
additional data for chemical interactions and flexible bond torsion positions.
They may be edited by the user.
Atom Definition Rules
The definition
files use a consistent atom labeling convention for which an atom in virtually
any chemical environment can be identified. The specification of adjacent atoms
is nested using the elements listed below.
·
Each element
must be separated by a space.
·
If
more than one adjacent atom is specified, then ALL must be present (i.e. a boolean AND for rules within a
line).
·
If a
label can have multiple definition lines, then any ONE of them must be
satisfied for inclusion (i.e. a boolean
OR for rules on different lines).
Atom Definition Elements
|
Element |
Function |
|
atom type |
Specifies partial or
complete atom type. A partial specification is more general (i.e.
"C" versus "C.3"). An asterisk (*) specifies ANY atom
type. |
|
( ) |
Specifies atoms that
must be bonded to parent atom. |
|
[ ] |
Specifies atoms that
must NOT be bonded to parent atom. |
|
integer |
Specifies the number
of an atom that must be bonded. |
Example Definitions
|
Example |
Explanation |
|
C.2 ( 2 O.co2 ) |
A carboxylate
carbon. |
|
.3 [ 3 H ] |
Any sp3 hybridized
atom that is not attached to three hydrogens. |
|
C. [
O. ] [ N. [ 2 O.2 ] [ 2 C. ] ] |
Any carbon not
attached to an oxygen or a nitrogen (unless the nitrogen
is a nitro or tertiary nitrogen). |
vdw.defn
This file contains atom labels and
definitions for van der Waals
atom typing.
·
The
following data types are associated with each atom: VDW radius, VDW well-depth,
flag for heavy atom, number of attached atoms.
·
Some
labels are used only for the united-atom model, some for only the all-atom
model, and some for either.
·
A
label may have multiple definitions.
Sample Entries
from vdw.defn
_____________________________________
name Carbon_sp/sp2
atom_model either
radius 1.850
well_depth 0.120
heavy_flag 1
valence 4
definition C
_____________________________________
name Carbon_All_sp3
atom_model all
radius 1.800
well_depth 0.060
heavy_flag 1
valence 4
definition C.3
_____________________________________
name Carbon_United_CH3
atom_model united
radius 2.000
well_depth 0.150
heavy_flag 1
valence 4
definition C. ( 3 H )
_____________________________________e
chem.defn
This file contains labels and definitions
for chemical labeling.
·
Nothing
in addition to a label is assigned to an atom.
·
A
label may have multiple definition lines.
Sample Entries
from chem.defn
________________________________________________________
name hydrophobic
definition C. [ O. ] [ N.
[ 2 O.2 ] [ 2 C. ] ] ( * )
definition N.pl3 ( 3 C. )
definition Cl ( C. )
definition Br ( C. )
definition I ( C. )
definition C.3 [ * ]
________________________________________________________
name donor
definition N. ( H )
definition N.4 [ * ]
________________________________________________________
name acceptor
definition O. [ H ] [ N. ] ( * )
definition O.3 ( 1 * ) [ N. ]
definition O.co2 ( C.2 ( O.co2 ) )
definition N. [ H ] [ N. ] [ O.
] [ 3 . ] ( * )
definition O.2 [ * ]
________________________________________________________
chem_match.tbl
This file contains the interaction matrix
for which chemical labels can form an interaction in matching.
·
The
labels must be identical to labels in chem.defn.
·
The
table flag indicates the beginning of the interaction table.
·
Compatible
labels are identified with a one, otherwise a zero.
Sample of chem_match.tbl
label null
label hydrophobic
label donor
label acceptor
label polar
table
1
1 1
1 0 1
1 0 0 1
1 0 1 1
1
nual Parameter Files
flex.defn
This file contains labels and definitions
for flexible bond identification.
·
The drive_id field corresponds to a torsion type in the flex_drive.tbl file.
·
The
minimize field is a flag for whether the bond may be minimized.
·
Two
definition lines must be present. Each definition corresponds to an atom at
either end of the bond.
Sample Entries from flex.defn
________________________________________
name sp3-sp3
drive_id 3
minimize 1
definition .3 [ 3 H ] [ 3 O.co2 ]
definition .3 [ 3 H ] [ 3 O.co2 ]
________________________________________
name sp3-sp2
drive_id 4
minimize 1
definition .3 [ 3 H ] [ 3 O.co2 ]
definition .2 [ 2 H ] [ 2 O.co2 ]
________________________________________
name sp2-sp2
drive_id 2
minimize 0
definition .2 [ 2 H ] [ 2 O.co2 ]
definition .2 [ 2 H ] [ 2 O.co2 ]
________________________________________
flex_drive.tbl
This file contains torsion positions
assigned to each rotatable bond when the flexible docking parameter
is used in DOCK.
·
The drive_id field corresponds to each torsion type.
·
The
positions field specifies the number of torsion angles to sample.
·
The
torsions field specifies the angles that are sampled.
Sample Entries from flex_drive.defn
_________________________________________
drive_id 2
positions 2
torsions 0 180
_________________________________________
drive_id 3
positions 3
torsions -60 60 180
_________________________________________
drive_id 4
positions 4
torsions -90 0 90 180
_________________________________________
APPENDIX 2: Molecular File Formats
Tripos MOL2 format
Overview
This format is
used for general molecule input and output of DOCK. This format has the advantage of storing all
the necessary information for atom features, position, and connectivity. It is
also a standardized format that other modeling programs can read.
Specification
Of the many
record types in a MOL2 file, DOCK recognizes the following: MOLECULE, ATOM,
BOND, SUBSTRUCTURE and SET. In the MOLECULE record, DOCK utilizes information
about the molecule name and number of atoms, bonds, substructures and sets. In
the ATOM record DOCK utilizes information about the atom names, types,
coordinates, and partial charges. In the BOND record, DOCK utilizes the atom
identifiers for the bond. In the SUBSTRUCTURE record, DOCK records the fields,
but does not utilize them. The SET records are entirely optional. They are used
only in special circumstances, like when
the use wants to define the anchor for flexible docking.
Please refer
to Sybyl documentation for additional information.
Example
This example file illustrates all the
elements of the MOL2 file read and written by dock. It includes optional SET
records which are used by the ligand flexibility routines.
@<TRIPOS>MOLECULE
example
23 23 1 0
2
GAST_HUCK
@<TRIPOS>ATOM
1 C 1.2300 0.7100
0.0000 C.ar 1 ABC -0.0529
2 H
2.1620 1.2490
0.0000 H 1 ABC 0.0571
3 C 0.0000 1.4200
0.0000 C.ar 1 ABC -0.0242
4 C 0.0000 2.9060
0.0000 C.3 1 ABC 0.0319
5 H -0.5150 3.2700
0.8920 H 1 ABC 0.0598
6 H -0.5150 3.2700
-0.8920 H 1 ABC 0.0598
7 N 1.3680 3.3900
0.0000 N.3 1 ABC -0.3075
8 H 1.8480 3.0500
0.8320 H 1 ABC
0.1243
9 C 1.3680 4.8410
0.0000 C.3 1 ABC -0.0136
10 H 0.8530 5.2050
0.8920 H 1 ABC 0.0390
11 H 0.8530 5.2050
-0.8920 H 1 ABC 0.0390
12 H 2.3990 5.2050
0.0000 H 1 ABC 0.0390
13 C -1.2300 0.7100
0.0000 C.ar 1 ABC -0.0529
14 H -2.1620 1.2490
0.0000 H 1 ABC 0.0571
15 C
-1.2300 -0.7100
0.0000 C.ar 1 ABC -0.0471
16 H -2.1620 -1.2490
0.0000 H 1 ABC 0.0655
17 C -0.0000 -1.4200
0.0000 C.ar 1 ABC 0.0547
18 C 1.2300 -0.7100
0.0000 C.ar 1 ABC -0.0471
19 H 2.1620 -1.2490
0.0000 H 1 ABC 0.0655
20 C -0.0000 -2.7800
0.0000 C.2 1 ABC 0.2555
21 O -0.0850 -3.5030
0.9750 O.2 1 ABC -0.3604
22 O 0.1090 -3.2660
-1.2480 O.3 1 ABC -0.2944
23 H 0.1010 -4.2400
-1.1490 H 1 ABC 0.2522
@<TRIPOS>BOND
1
1 2
1
2
1 18 ar
3 1 3 ar
4
3 13 ar
5
3 4 1
6
4 6 1
7
4 5 1
8
4 7 1
9
7 8 1
10
7 9 1
11
9 12 1
12
9 11 1
13
9 10 1
14
13 14 1
15
13 15 ar
16
15 16 1
17
15 17 ar
18
17 20 1
19
17 18 ar
20
18 19 1
21
20 21 2
22
20 22 1
23
22 23 1
@<TRIPOS>SUBSTRUCTURE
1 ABC
1 GROUP 0 A ****
0 ROOT
@<TRIPOS>SET
ANCHOR STATIC ATOMS
<user> **** Anchor atom
set
6 1 3 13 15 17 18
RIGID STATIC BONDS
<user> **** Rigid bond set
6 2 3 4 15 17 19