Amber Score in DOCK6
Author: Devleena Shivakumar
Last updated June 6, 2025 by Scott Brozell
This tutorial introduces DOCK Amber score and describes the preparation of input files for DOCK Amber rescoring. These techniques should be applicable to any protein-ligand system. Docking and rescoring studies of 3,5 difluoroaniline to M102Q/L99A T4 Lysozyme Protein (PDB ID 1LGU ) are the basis; see J. Mol. Biol. 377, 914-934 (2008).
What is Amber score?
The generalized Born/surface area (GB/SA) continuum model for solvation free energy is a fast and accurate alternative to an explicit solvent model for molecular simulations. We have implemented this physics-based method in the Amber scoring function in the program DOCK6. To curtail the computational cost while still maintaining accuracy, the atoms distant from the site of the ligand binding can be kept frozen. Thus, CPU time is not spent updating those energies and derivatives during the course of the simulation. The main advantage of Amber score is that both the ligand and the active site(s) of the receptor can be flexible, allowing small structural rearrangements to reproduce the so-called "induced fit" while performing the scoring.
Amber score performs minimization, molecular dynamics (MD) simulation, and more minimization on the individual ligand, the individual receptor, and the ligand-receptor complex, and calculates the score as follows:
Ebinding = Ecomplex - (Ereceptor + Eligand)
where E is obtained from:
E = EMM + (Ep-sol + Enp-sol)
EMM = Ebad + EvdW + Ees --- AMBER MM force field
Ep-sol --- Electrostatic part of solvation energy using GB
Enp-sol --- Non-polar part of solvation energy using SA
The user has the option to increase or decrease the number of minimization and MD simulation steps. However, it may not be desirable to have a large number of steps due to the time taken for the calculations. For various protein test cases, we have found 100 minimization and 3000 MD steps to produce good results. These are set as defaults in the program.
Writing the topmost poses from a DOCK calculation with a less expensive score than Amber score is highly recommended. Then Amber score should be used on these topmost poses for each ligand. For example, for T4 Lysozyme the DOCK score is calculated for 1 million compounds from the Available Chemicals Directory (ACD). The top 5000-10,000 compounds ranked by DOCK are passed through Amber score for further refinement. This is further illustrated in the cartoon below:
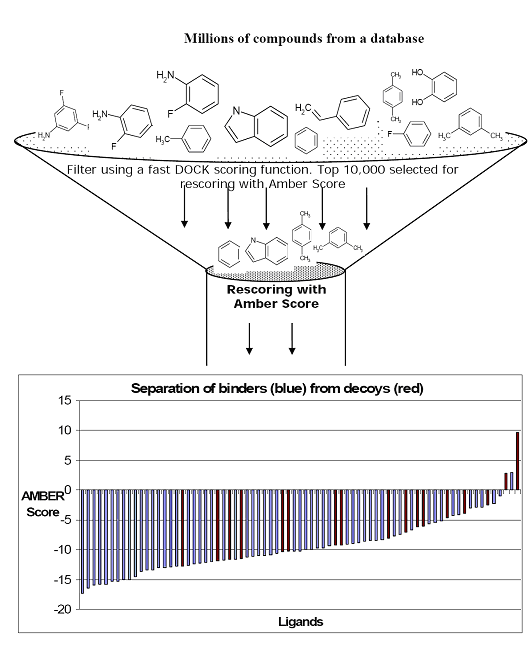
Input files preparation.
1) Ligand Preparation: Start with the output scored mol2 file from a previous DOCK run: lig.mol2.
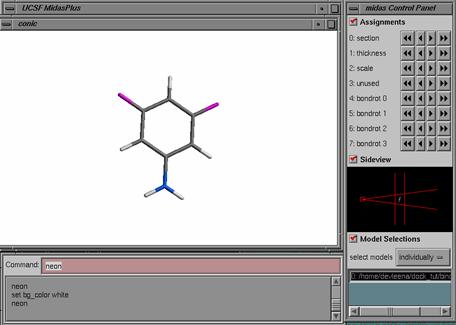
lig.mol2 is the topmost pose generated for the ligand 3,5 difluoroaniline from a previous DOCK run. Also notice that this mol2 file is an output from DOCK, therefore the atom types are in SYBYL file format. For Amber score, we need the atom types in GAFF file format which is compatible with the AMBER molecular mechanics force field. You'll need a script prepare_amber.pl that will do the trick for you. If you have installed DOCK6, this script can be found in the bin directory.
2) Receptor Preparation:
(a) Clean the PDB file:
Download the receptor PDB file 1lgu.pdb.
Remove all the ligand atoms, ions, and crystal water molecules from the receptor pdb file. If you know that certain water molecules or ions play catalytic or structural roles, use your scientific judgment to decide whether to keep them in the PDB file.
(b) Determine the protonation state of titratable groups:
Determine the protonation state of the histidine and other titratable residues in the receptor. Care should be taken to assign the appropriate protonation state, especially if the residue is at or near the active site or within the flexible region of the scoring calculation. Use experimental data from the literature whenever available, or your chemical intuition to assign the protonation states for these residues. [Hint: Check for hydrogen bonding residues nearby to see whether His or Asp should be protonated.] Or, you can use softwares to do this job. Some examples:
i. PDB2PQR is a Python software package that automates the PDB file preparation and protonation state assignments.
ii. H++ is a tool to estimate pKa's of protein side chains, and to automate the process of assigning protonation states for molecular dynamics simulations.
(c) Verify the residue names in the PDB file:
After assigning the protonation states, make sure that your receptor PDB file has residue names according to the Amber readable format. Check the name of the residues to ensure that they have the correct names:
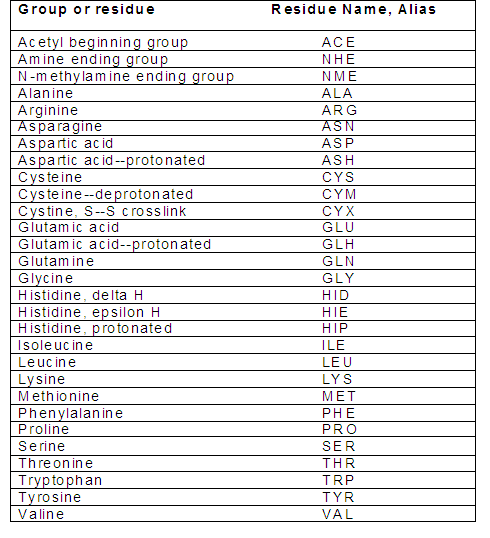
Note that in Amber, histidine has three different residue names based on the location of the proton(s) in the imidazole ring. The default residue name in the PDB file from RCSB is HIS. If you do not rename this, the program assumes that it is imidazole with a proton on the N-epsilon atom, and assigns it residue name HIE. In addition, if disulfide bonds are important then verify that the pdb file contains correct conect records for them, and change the residue names from CYS to CYX.
3) Generation of Amber readable input files:
Prepare Amber readable input files for each receptor, ligand and the corresponding complex. This is done with the help of a perl script that is provided in the bin directory. (Note that the environment variable AMBERHOME should be undefined when using this script.) To run this script, first verify that you have perl installed on your machine. Type the following at the command prompt:
$ which perl
You should get an output similar to
/usr/bin/perl
If you cannot find perl then please install it.
The syntax for prepare_amber.pl is
USAGE: prepare_amber.pl ligand_mol2_file receptor_PDB_file
For this example:
prepare_amber.pl lig.mol2 1lgu.pdb
The output files are
Files associated with the receptor
amberize_receptor.out
1lgu.amber.pdb
1lgu.inpcrd
1lgu.log
1lgu.prmtop
Files associated with the ligand:
amberize_ligand.lig.1.out
lig.amber_score.mol2
lig.1.amber.pdb
lig.1.frcmod
lig.1.gaff.mol2
lig.1.inpcrd
lig.1.log
lig.1.mol2
lig.1.mopac.out
lig.1.prmtop
Files associated with the complex:
amberize_complex.lig.1.out
1lgu.lig.1.amber.pdb
1lgu.lig.1.inpcrd
1lgu.lig.1.log
1lgu.lig.1.prmtop
prepare_amber.pl has the capability to read and process a single file containing multiple ligands - typically what you would get from a DOCK run. It splits a file containing multiple mol2 entries into individual mol2 files and produces the files with extensions .amber.pdb, .inpcrd, and .prmtop that are later read by the Amber score. Since in this example, there was only one ligand in lig.mol2, the mol2 output was lig.1.mol2. Had there been 3 ligands in a ligand_mol2_file named mydnagil.mol2, the mol2 outputs would have been: mydnagil.1.mol2, mydnagil.2.mol2, and mydnagil.3.mol2.
The script prepare_amber.pl performs the following main tasks:
(i) Reads the PDB file for the receptor; adds hydrogens and some other
missing atoms if not present; adds AMBER force field atom types and
charges. Generates Amber parameter/topology (prmtop) and coordinate
(inpcrd) files. This is performed via the script amberize_receptor which
invokes program tleap.
(ii) Generates a modified mol2 file with suffix amber_score.mol2 (e.g.,
lig.amber_score.mol2) that appends to each TRIPOS MOLECULE a TRIPOS
AMBER_SCORE_ID section containing a unique label (e.g., lig.1,
mydnagil.1, etc.). This modified mol2 file should be assigned to the
ligand_atom_file input parameter in the DOCK input file.
(iii) Splits the ligand_mol2_file into separate mol2 files, one file per
ligand, where the file prefixes are the AMBER_SCORE_ID unique labels.
These mol2 files are used in the next step.
(iv) Runs the antechamber program to determine the semi-empirical
charges, such as AM1-BCC, for each ligand. Runs the tleap program to
create parameter/topology and coordinate files for each ligand using the
GAFF force field (prmtop, frcmod, and inpcrd), and writes a mol2 file
for the ligands with GAFF atom types (gaff.mol2). This is performed via
the script amberize_ligand.
(v) Combines each ligand with the receptor to generate the
parameter/topology and coordinate files for each complex. This is
performed via the script amberize_complex which invokes program tleap.
The script prepare_amber.pl also creates files with extensions .log and .out that contain the detailed results of the above steps. The amberize_*.out files can be browsed to verify that the preparation was successful. If prepare_amber.pl emits any warnings or errors then all these files should be scrutinized. Some errors are fatal, such as an inability to prepare the receptor; generally, and in this case especially start investigating by examining file amberize_receptor.out. Since preparation for Amber score is more stringent than preparation for other DOCK scores, usually a small fraction of a large set of ligands fails during prepare_amber.pl execution. This Dock-fans post discusses in detail the recovery options for failing ligands.
4) Creation of a DOCK6 input file for Amber Score:
Prepare an input file (dock.in) for DOCK6 or download it:
dock.in.
The following options are Amber score specific:
ligand_atom_file............................. lig.amber_score.mol2
amber_score_primary.......................... yes
amber_score_receptor_file_prefix............. 1lgu
amber_score_movable_region................... ligand
amber_score_minimization_rmsgrad............. 0.01
amber_score_before_md_minimization_cycles.... 100
amber_score_md_steps......................... 3000
amber_score_after_md_minimization_cycles..... 100
amber_score_gb_model......................... 5
amber_score_nonbonded_cutoff................. 18.0
amber_score_temperature...................... 300.0
amber_score_abort_on_unprepped_ligand........ yes
For ligand_atom_file, use the output file with the suffix .amber_score.mol2 generated by prepare_amber.pl (see (ii) above).
Use the prefix of the receptor PDB file as the input for amber_score_receptor_file_prefix. For this example amber_score_receptor_file_prefix is 1lgu for the pdb file 1lgu.pdb.
Choose amber_score_movable_region as ligand. This defines the region that is allowed to move during scoring. To select other options, please read the DOCK 6 Manual
Run DOCK6 with Amber Score.
USAGE: dock6 -i input_file [-o output_file] [-v]
dock6 -i dock.in > dock.out &
Download a copy of the dock.out file. Check it with your output file to see whether you get any different results. Since this is a rescoring calculation of a single ligand, the DOCK _scored.mol2 file contains only that single ligand: output_scored.mol2. For Amber score this file contains the ligand coordinates from the final pose of the complex, the ligand charges from the ligand prmtop file, and the ligand DOCK atom types (not the GAFF atom types used by Amber score). Since the ligand was movable two additional output files are produced: lig.1.final_pose.amber.pdb and 1lgu.lig.1.final_pose.amber.pdb. These contain the final poses of the ligand and the complex, respectively, after the Amber score protocol.
You can now change the options in the dock.in file, such as increase the number of steps of the amber_score minimization cycles and/or amber_score_md_steps, or increase/decrease the amber_score_temperature, change the amber_score_movable_region, and rerun the calculation to see the change in results. In addition, try rerunning with verbose output via the -v flag.
In general, the best type of movable region could be highly sensitive to the receptor. For example, if specific changes to the receptor upon ligand binding are expected then the NAB_ATOM_EXPRESSION movable region is appropriate. On the other hand, if a little relaxation of the receptor upon ligand binding is all that is desired then a small DISTANCE movable region might be sufficient.
The amount of minimization and MD is dependent on the preparation of the receptor. If the receptor has been well equilibrated then it is expected that fewer min. steps and MD cycles will be needed to relax the complex, in general. Other factors, such as the type and extent of changes upon binding, are also relevant.